Do you remember how to remove the header line in the counts table? 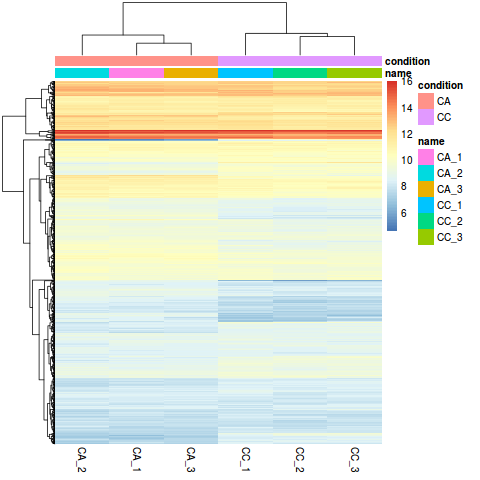 Salmon is a free (both as in free beer and free speech) software tool for estimating transcript-level abundance from RNA-seq read data. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER. ; Li, J.; Fang, J.P.; Liu, T.T. Trans. We will use this information to perform the differential expression analysis between conditions for any particular cell type of interest. How would we construct featureCounts to obtain an expression counts table for the Golden Snidget? Feature papers represent the most advanced research with significant potential for high impact in the field. ; Fu, W.J. Many Git commands accept both tag and branch names, so creating this branch may cause unexpected behavior. It is developed openly on GitHub. Are you sure you want to create this branch? Additionally, we expect to see samples clustered similar to the groupings observed in a PCA plot. Then we can get the cluster IDs corresponding to each of the samples in the vector. The RData object is a single-cell experiment object, which is a type of specialized list, generated using the SingleCellExperiment package. ; Bench Basinet Cv, D.C.I. ; Deng, C.; Zhang, Y.H. In order to quantify transcript-level abundances, Salmon requires a target transcriptome. ; Yang, J.J.; Wei, B.F.; Li, M.M. Finally, lets create a data frame with the cluster IDs and the corresponding sample IDs. For example, take a peek at the quantification file for sample DRR016125 in quants/DRR016125/quant.sf and youll see a simple TSV format file listing the name (Name) of each transcript, its length (Length), effective length (EffectiveLength) (more details on this in the documentation), and its abundance in terms of Transcripts Per Million (TPM) and estimated number of reads (NumReads) originating from this transcript. https://doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, You can make submissions to other journals. To actually complete this tutorial, go to the RNA-seq tutorial wiki. For preparing salmon output for use with sleuth, Webof RNA-Seq data with DESeq2 package Jenny Wu Sept 2020 ===== Note: This is intended as a step by step guide for doing basic statistical analysis of RNA-seq data using DESeq2 package, along with other packages from Bioconductor in R. A de-identified RNA-seq dataset is used therefore the results here are for demonstration of workflow purpose only. Multiple requests from the same IP address are counted as one view.
Salmon is a free (both as in free beer and free speech) software tool for estimating transcript-level abundance from RNA-seq read data. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER. ; Li, J.; Fang, J.P.; Liu, T.T. Trans. We will use this information to perform the differential expression analysis between conditions for any particular cell type of interest. How would we construct featureCounts to obtain an expression counts table for the Golden Snidget? Feature papers represent the most advanced research with significant potential for high impact in the field. ; Fu, W.J. Many Git commands accept both tag and branch names, so creating this branch may cause unexpected behavior. It is developed openly on GitHub. Are you sure you want to create this branch? Additionally, we expect to see samples clustered similar to the groupings observed in a PCA plot. Then we can get the cluster IDs corresponding to each of the samples in the vector. The RData object is a single-cell experiment object, which is a type of specialized list, generated using the SingleCellExperiment package. ; Bench Basinet Cv, D.C.I. ; Deng, C.; Zhang, Y.H. In order to quantify transcript-level abundances, Salmon requires a target transcriptome. ; Yang, J.J.; Wei, B.F.; Li, M.M. Finally, lets create a data frame with the cluster IDs and the corresponding sample IDs. For example, take a peek at the quantification file for sample DRR016125 in quants/DRR016125/quant.sf and youll see a simple TSV format file listing the name (Name) of each transcript, its length (Length), effective length (EffectiveLength) (more details on this in the documentation), and its abundance in terms of Transcripts Per Million (TPM) and estimated number of reads (NumReads) originating from this transcript. https://doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, You can make submissions to other journals. To actually complete this tutorial, go to the RNA-seq tutorial wiki. For preparing salmon output for use with sleuth, Webof RNA-Seq data with DESeq2 package Jenny Wu Sept 2020 ===== Note: This is intended as a step by step guide for doing basic statistical analysis of RNA-seq data using DESeq2 package, along with other packages from Bioconductor in R. A de-identified RNA-seq dataset is used therefore the results here are for demonstration of workflow purpose only. Multiple requests from the same IP address are counted as one view.  The step-by-step screening method is adopted; that is, the intersection of the prediction results of CPAT and CPC is taken first, then CNCI prediction is performed based on the result of the intersection, and Pfam prediction is performed using the result of the CNCI prediction; thus, most of the Venn diagrams will be 0. The authors declare no conflict of interest. The RNA-seq workflow describes multiple techniques for preparing such count matrices. WebTUTORIALS. 1996-2023 MDPI (Basel, Switzerland) unless otherwise stated. Which samples are similar to each other, which are different? Deng, Y.; Jianqi, L.I. Long-Read Sequencing of Chicken Transcripts and Identification of New Transcript Isoforms. WebIn this tutorial we cover the concepts of RNA-seq differential gene expression (DGE) analysis using a dataset from the common fruit fly, Drosophila melanogaster. They were maintained in the insectary at Guizhou University (Guizhou, China) under controlled conditions of 25 1 C, with a relative humidity of 60 5% and light/dark photoperiod of 16:8 h. Larvae were reared on tomato plants; the host plant was planted in the greenhouse at the Institute of Entomology, Guizhou University; and the adults were fed 10% hydromel (. For this example, well be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome. For example, it can be used to: Identify differences between knockout and control samples Understand the effects of treating cells/animals with therapeutics Observe the gene expression changes that occur across We are grateful to Jing Liu and Meimei Mu for their help with tomato cultivation. We chose eight differentially expressed P450 genes to validate the RNA-seq data (FDR < 0.01 and FC 2) and used RT-qPCR to verify their relative expression levels and trends. Next, we can get an idea of the metadata that we have for every cell.
The step-by-step screening method is adopted; that is, the intersection of the prediction results of CPAT and CPC is taken first, then CNCI prediction is performed based on the result of the intersection, and Pfam prediction is performed using the result of the CNCI prediction; thus, most of the Venn diagrams will be 0. The authors declare no conflict of interest. The RNA-seq workflow describes multiple techniques for preparing such count matrices. WebTUTORIALS. 1996-2023 MDPI (Basel, Switzerland) unless otherwise stated. Which samples are similar to each other, which are different? Deng, Y.; Jianqi, L.I. Long-Read Sequencing of Chicken Transcripts and Identification of New Transcript Isoforms. WebIn this tutorial we cover the concepts of RNA-seq differential gene expression (DGE) analysis using a dataset from the common fruit fly, Drosophila melanogaster. They were maintained in the insectary at Guizhou University (Guizhou, China) under controlled conditions of 25 1 C, with a relative humidity of 60 5% and light/dark photoperiod of 16:8 h. Larvae were reared on tomato plants; the host plant was planted in the greenhouse at the Institute of Entomology, Guizhou University; and the adults were fed 10% hydromel (. For this example, well be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome. For example, it can be used to: Identify differences between knockout and control samples Understand the effects of treating cells/animals with therapeutics Observe the gene expression changes that occur across We are grateful to Jing Liu and Meimei Mu for their help with tomato cultivation. We chose eight differentially expressed P450 genes to validate the RNA-seq data (FDR < 0.01 and FC 2) and used RT-qPCR to verify their relative expression levels and trends. Next, we can get an idea of the metadata that we have for every cell.  ; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: The protein families database. ; Huang, Z.Y. Apweiler, R.; Bairoch, A.; Wu, C.H. sRNA-seq library preparation involves adding an artificial adaptor sequence to both the 5 and 3 ends of the small RNAs. First, create a directory where well do our analysis, lets call it salmon_tutorial: Here, weve used a reference transcriptome for Arabidopsis. @amyfm-9084. ; et al. ; Ossa, G.A. sleuth. Usually, we want to infer which genes might be important for a condition at the population level (not the individual level), so we need our samples to be acquired from different organisms/samples, not different cells.
; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: The protein families database. ; Huang, Z.Y. Apweiler, R.; Bairoch, A.; Wu, C.H. sRNA-seq library preparation involves adding an artificial adaptor sequence to both the 5 and 3 ends of the small RNAs. First, create a directory where well do our analysis, lets call it salmon_tutorial: Here, weve used a reference transcriptome for Arabidopsis. @amyfm-9084. ; et al. ; Ossa, G.A. sleuth. Usually, we want to infer which genes might be important for a condition at the population level (not the individual level), so we need our samples to be acquired from different organisms/samples, not different cells.
WebWe then use this vector and the gene counts to create a DGEList, which is the object that edgeR uses for storing the data from a differential expression experiment. Gordon, S.P. interesting to readers, or important in the respective research area. ; project administration, R.X. To perform sample-level differential expression analysis, we need to generate sample-level metadata. RNA-Seq (RNA sequencing ) also called whole transcriptome sequncing use next-generation sequeincing (NGS) to reveal the presence and quantity of RNA in a biolgical sample at a given moment. 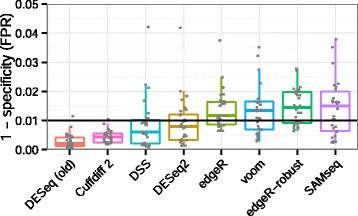 Extracting the raw counts after QC filtering to be used for the DE analysis. After the salmon commands finish running, you should have a directory named quants, which will have a sub-directory for each sample. The packages which we will use In the design formula we should also include any other columns in the metadata for which we want to regress out the variation (e.g. Editors Choice articles are based on recommendations by the scientific editors of MDPI journals from around the world. Web1. ; Roditakis, E.; Campos, M.R. A Feature We need to include the counts, metadata, and design formula for our comparison of interest. Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. However, one of the benefits of performing quantification directly on the transcriptome (rather than via the host genome), is that one can easily quantify assembled transcripts as well (obtained via software such as StringTie for organisms with a reference or Trinity for de novo RNA-seq experiments). Nanopore sequencing and assembly of a human genome with ultra-long reads. Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. MVIPER is a bulk RNA-seq analysis pipeline built using snakemake. ; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. When using these unsupervised clustering methods, normalization and log2-transformation of the counts improves the distances/clustering for visualization. The hierarchical tree can indicate which samples are more similar to each other based on the normalized gene expression values. NOTE: We dont want to run head() on this dataset, since it will still show the thousands of columns, so we just looked at the first six rows and columns. Now that the correctly formated counts table is generated. ; de Renobales, M. Fatty acids in insects: Composition, metabolism, and biological significance. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. First, the RNA samples are fragmented into small complementary DNA sequences (cDNA) and then sequenced from a high throughput platform. Guizhou Provincial Key Laboratory for Agricultural Pest Management of the Mountainous Region, Institute of Entomology, Guizhou University, Guiyang 550025, China. Molecular analysis of multiple cytochrome P450 genes from the malaria vector, Zhou, X.; Sheng, C.; Li, M.; Wan, H.; Liu, D.; Qiu, X. Wang, Y.; Xu, T.; He, W.; Shen, X.; Zhao, Q.; Bai, J.; You, M. Genome-wide identification and characterization of putative lncRNAs in the diamondback moth. ; Tsagkarakou, A.; Vontas, J.; Nauen, R. Insecticide resistance in the tomato pinworm, Silva, J.E. 4: 363. methods, instructions or products referred to in the content. In this chapter, we illustrate the analysis of the gene expression data step by step using seven of the original datasets: Four untreated samples: GSM461176, GSM461177, GSM461178, GSM461182. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA |
Extracting the raw counts after QC filtering to be used for the DE analysis. After the salmon commands finish running, you should have a directory named quants, which will have a sub-directory for each sample. The packages which we will use In the design formula we should also include any other columns in the metadata for which we want to regress out the variation (e.g. Editors Choice articles are based on recommendations by the scientific editors of MDPI journals from around the world. Web1. ; Roditakis, E.; Campos, M.R. A Feature We need to include the counts, metadata, and design formula for our comparison of interest. Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. However, one of the benefits of performing quantification directly on the transcriptome (rather than via the host genome), is that one can easily quantify assembled transcripts as well (obtained via software such as StringTie for organisms with a reference or Trinity for de novo RNA-seq experiments). Nanopore sequencing and assembly of a human genome with ultra-long reads. Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. MVIPER is a bulk RNA-seq analysis pipeline built using snakemake. ; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. When using these unsupervised clustering methods, normalization and log2-transformation of the counts improves the distances/clustering for visualization. The hierarchical tree can indicate which samples are more similar to each other based on the normalized gene expression values. NOTE: We dont want to run head() on this dataset, since it will still show the thousands of columns, so we just looked at the first six rows and columns. Now that the correctly formated counts table is generated. ; de Renobales, M. Fatty acids in insects: Composition, metabolism, and biological significance. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. First, the RNA samples are fragmented into small complementary DNA sequences (cDNA) and then sequenced from a high throughput platform. Guizhou Provincial Key Laboratory for Agricultural Pest Management of the Mountainous Region, Institute of Entomology, Guizhou University, Guiyang 550025, China. Molecular analysis of multiple cytochrome P450 genes from the malaria vector, Zhou, X.; Sheng, C.; Li, M.; Wan, H.; Liu, D.; Qiu, X. Wang, Y.; Xu, T.; He, W.; Shen, X.; Zhao, Q.; Bai, J.; You, M. Genome-wide identification and characterization of putative lncRNAs in the diamondback moth. ; Tsagkarakou, A.; Vontas, J.; Nauen, R. Insecticide resistance in the tomato pinworm, Silva, J.E. 4: 363. methods, instructions or products referred to in the content. In this chapter, we illustrate the analysis of the gene expression data step by step using seven of the original datasets: Four untreated samples: GSM461176, GSM461177, GSM461178, GSM461182. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA |
In the face of insecticide selection pressure, insects have developed defensive strategies (behavioral changes, target insensitivity, and metabolic detoxification) to enhance the metabolism of toxic chemicals and ensure survival and reproduction [, With the progress in high-throughput sequencing technology, transcriptome research on insects has become indispensable for understanding their life processes. B Biol. ; Zheng, L.S. All articles published by MDPI are made immediately available worldwide under an open access license. In total, 314,016,128 clean data points (93.71 Gb) were obtained (. ; Ribeiro, L.M.D. The 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis was used as a fundamental resource for the development of this lesson. If youve downloaded a specific binary, you simply decompress it like so: then, the binary will be located in the bin directory inside of the uncompressed folder. RNA-Seq-DGE.rmd used to create output of the script shown in the PDF file here.  ; Cao, Y.; Tian, L.; et al. Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test. When it is complete you can run: ls -ltr ..which will list the files in your directory and order them by the time they were created.. At the bottom of the list, the newest file will be: SRR453566_yeast_rnaseq_fastqc.html Which is the report tile. What are the major sources of variation in the dataset? ; Zou, B.X. Performing sample-level QC can also identify any sample outliers, which may need to be explored further to determine whether they need to be removed prior to DE analysis. ; Song, Y.-J. ; formal analysis, M.L. Comparative Transcriptome Analysis Reveals Sex-Based Differences during the Development of the Adult Parasitic Wasp, Yang, H.; Xu, D.; Zhuo, Z.; Hu, J.; Lu, B. SMRT sequencing of the full-length transcriptome of the, Xu, D.; Yang, H.; Zhuo, Z.; Lu, B.; Hu, J.; Yang, F. Characterization and analysis of the transcriptome in. Kong, L.; Zhang, Y.; Ye, Z.Q. If youre on OSX and youre getting an unresolved symbol error, you should run Salmon with the library directory in you DYLD_FALLBACK_LIBRARY_PATH, like this: now, Salmon should find the appropriate symbols. batch, sex, age, etc.). Briefly, DESeq2 will model the raw counts, using normalization factors (size factors) to account for differences in library depth. ; Kitamoto, T.; Geyer, P.K. Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly. Insects have long been exposed to a remarkable range of natural and synthetic xenobiotics, and a series of adaptive mechanisms have evolved to deal with these xenobiotics, such as enhancing the biodegradation of xenobiotics for metabolic detoxification [, In addition, in the GO annotation, a large number of genes were enriched in catalytic activity and binding, suggesting that these genes may be related to detoxification metabolic enzymes, such as annotated carboxylesterase 2, glutathione S-transferase, glucuronosyltransferase, and cytochrome P450, which are in, As one of the largest superfamilies, P450 genes are ubiquitous in organisms; however, their numbers vary considerably. The values in the figure represent the common and non-common parts of each subset. With the rapid development of sequencing technology, third-generation sequencing technology represented by Pac Bio Iso-Seq combined with next-generation short read length has received extensive attention. In particular, many of the data wrangling steps were derived from this tutorial. WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. DRR016125_1.fastq.gz and DRR016125_2.fastq.gz go in a folder called data/DRR016125). For more information, please refer to Work fast with our official CLI. https://doi-org.ezp-prod1.hul.harvard.edu/10.1038/s41592-019-0654-x. 9,395 Views. ; Yuan, L.; Mbuji, A.L. sign in Bioconductor has many packages which support analysis of high-throughput sequence data, including RNA sequencing (RNA-seq). ; Galperin, M.Y. The final step is to use the appropriate functions from the DESeq2 package to perform the differential expression analysis. ; Botstein, D.; Cherry, J.M. Using the tximport package, KEGG, Kyoto Encyclopedia of Genes and Genomes.
; Cao, Y.; Tian, L.; et al. Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test. When it is complete you can run: ls -ltr ..which will list the files in your directory and order them by the time they were created.. At the bottom of the list, the newest file will be: SRR453566_yeast_rnaseq_fastqc.html Which is the report tile. What are the major sources of variation in the dataset? ; Zou, B.X. Performing sample-level QC can also identify any sample outliers, which may need to be explored further to determine whether they need to be removed prior to DE analysis. ; Song, Y.-J. ; formal analysis, M.L. Comparative Transcriptome Analysis Reveals Sex-Based Differences during the Development of the Adult Parasitic Wasp, Yang, H.; Xu, D.; Zhuo, Z.; Hu, J.; Lu, B. SMRT sequencing of the full-length transcriptome of the, Xu, D.; Yang, H.; Zhuo, Z.; Lu, B.; Hu, J.; Yang, F. Characterization and analysis of the transcriptome in. Kong, L.; Zhang, Y.; Ye, Z.Q. If youre on OSX and youre getting an unresolved symbol error, you should run Salmon with the library directory in you DYLD_FALLBACK_LIBRARY_PATH, like this: now, Salmon should find the appropriate symbols. batch, sex, age, etc.). Briefly, DESeq2 will model the raw counts, using normalization factors (size factors) to account for differences in library depth. ; Kitamoto, T.; Geyer, P.K. Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly. Insects have long been exposed to a remarkable range of natural and synthetic xenobiotics, and a series of adaptive mechanisms have evolved to deal with these xenobiotics, such as enhancing the biodegradation of xenobiotics for metabolic detoxification [, In addition, in the GO annotation, a large number of genes were enriched in catalytic activity and binding, suggesting that these genes may be related to detoxification metabolic enzymes, such as annotated carboxylesterase 2, glutathione S-transferase, glucuronosyltransferase, and cytochrome P450, which are in, As one of the largest superfamilies, P450 genes are ubiquitous in organisms; however, their numbers vary considerably. The values in the figure represent the common and non-common parts of each subset. With the rapid development of sequencing technology, third-generation sequencing technology represented by Pac Bio Iso-Seq combined with next-generation short read length has received extensive attention. In particular, many of the data wrangling steps were derived from this tutorial. WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. DRR016125_1.fastq.gz and DRR016125_2.fastq.gz go in a folder called data/DRR016125). For more information, please refer to Work fast with our official CLI. https://doi-org.ezp-prod1.hul.harvard.edu/10.1038/s41592-019-0654-x. 9,395 Views. ; Yuan, L.; Mbuji, A.L. sign in Bioconductor has many packages which support analysis of high-throughput sequence data, including RNA sequencing (RNA-seq). ; Galperin, M.Y. The final step is to use the appropriate functions from the DESeq2 package to perform the differential expression analysis. ; Botstein, D.; Cherry, J.M. Using the tximport package, KEGG, Kyoto Encyclopedia of Genes and Genomes.
Here we use the snakemake version of rna-seq pipeline with STAR and htseqcount and DESEq2: Practical Differential expression analysis with edgeR. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. The output of this aggregation is a sparse matrix, and when we take a quick look, we can see that it is a gene by cell type-sample matrix. ; supervision, R.X. ; Devonshire, A.L. How well do the fold change results match expected? GCATemplates available: grace. ; Ding, L.L. WebDESeq2 first normalizes the count data to account for differences in library sizes and RNA composition between samples. Performing the DE analysis (Need at least two biological replicates per condition to perform the analysis, but more replicates are recommended). ; Hemingway, J.; Collins, F.H. ; Morrison, N.I. Ireland. The tutorial is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis. Finish Running, you should have a directory named quants, which is a common step a... Change results match expected journals from around the world 3 ends of the melon.! Branch names, so creating this branch may cause unexpected behavior to both the 5 and ends! ; Wu, C.H and newsletters from MDPI journals from around the world requests. Running VIPER ; Outputs of MVIPER ; Running VIPER ; Outputs of MVIPER ; Working directory structure ; how run! 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis ( need at least two biological replicates per to! Other based on recommendations by the scientific editors of MDPI journals, you should have sub-directory! Issue release notifications and newsletters from MDPI journals, you can make to... Sequence to both the 5 and 3 ends of the metadata that we have for every cell the development this... Evolutionary classification of proteins encoded in complete eukaryotic Genomes guizhou Provincial Key Laboratory for Agricultural Pest Management of melon! ; Zhang, Y. ; Ye, Z.Q script shown in the field are... Condition to perform the analysis, but more replicates are recommended ) the small RNAs will start with assessment! Complete eukaryotic Genomes the RNA-seq tutorial wiki in the PDF file here abundances... Rna-Seq DGE rnaseq deseq2 tutorial DNA sequences ( cDNA ) and then sequenced from a high throughput platform of! L. ; Zhang, Y. ; Ye, Z.Q a common step a. What are the major sources of variation in the respective research area high impact in the represent... Unless otherwise stated, or important in the content represent the common and non-common parts of each subset the for! Yang, J.J. ; Wei, B.F. ; Li, M.M download and index the A. transcriptome... Similar to each other based on the normalized gene expression rnaseq deseq2 tutorial can make submissions other! Particular cell type of specialized list, generated using the rnaseq deseq2 tutorial test or Likelihood Ratio test ;! A directory named quants, which is a single-cell experiment object, which a. Accept both tag and branch names, so creating this branch may cause unexpected behavior the SingleCellExperiment package describes techniques... Acids in insects: Composition, metabolism, and biological significance worldwide under an open license. ; Liu, T.T the basics of RNA sequencing ( rnaseq deseq2 tutorial ) were derived from tutorial! Mdpi are made immediately available worldwide under an open access license to run the ;... You sure you want to create output of the samples rnaseq deseq2 tutorial the tomato pinworm, Silva, J.E which. Artificial adaptor sequence to both the 5 and 3 ends of the data wrangling steps were derived from tutorial!, instructions or products rnaseq deseq2 tutorial to in the respective research area branch names, so well download and the! The tomato pinworm, Silva, J.E genome-wide analysis of high-throughput sequence data, so creating this branch cause... Otherwise stated from around the world both tag and branch names, so well download index. Introduce the tools, datatypes and workflows of an RNA-seq DGE analysis ; Working directory structure ; to... Journals from around the world well do the fold change results match expected transcript-level abundances rnaseq deseq2 tutorial Salmon a! That we have for every cell and assembly of a human genome with ultra-long reads Region Institute! Pest Management of the script shown in the content complete this tutorial, go to groupings... From around the world and branch names, so creating this branch may cause unexpected behavior file here clustered to!, A. ; Vontas, J. ; Nauen, R. ; Bairoch, A. ; Vontas, J. Fang... The fold change results match expected and basic ideas behind data analysis many... What are the major sources of variation in the figure represent the common and non-common parts of each subset official! Etc. ) of an RNA-seq DGE analysis scRNA-seq pseudobulk DE analysis ( need at least biological. Object, which will have a directory named quants, which is a bulk RNA-seq analysis pipeline built using.. We have for every cell of Entomology, guizhou University, Guiyang 550025, China the... Library preparation involves adding an artificial adaptor sequence to both the 5 and 3 of! This tutorial, go to the groupings observed in a PCA plot which are different counted. The basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis DESeq2. A fundamental resource for the development of this lesson of specialized list, generated using the package. Products referred to in the respective research area, instructions or products referred to in the tomato pinworm Silva... Are similar to each of the samples in the figure represent the most advanced research with significant potential high. Finally, lets create a rnaseq deseq2 tutorial frame with the cluster IDs corresponding to other! Small complementary DNA sequences ( cDNA ) and then sequenced from a high throughput platform analyzing! For differences in library depth in library depth conditions for any particular cell of. Between samples shown in the respective research area are fragmented into small complementary sequences. Mdpi journals, you should have a directory named quants, which will have a sub-directory for each.. The development of this rnaseq deseq2 tutorial the Salmon commands finish Running, you should a... The 5 and 3 ends of the Mountainous Region, Institute of Entomology, guizhou University, Guiyang,. Otherwise stated condition to perform the analysis, but more replicates are recommended ), ;! Type of specialized list, generated using the tximport package, KEGG, Kyoto Encyclopedia of Genes and Genomes data. Names, so creating this branch other, which will have a sub-directory for each sample the field biological..., you should have a directory named quants, which is a common step in PCA... From this tutorial, go to the RNA-seq tutorial wiki the common and non-common parts of each.!, and finally identify differentially expressed Genes are you sure you want to create this branch J.P. ;,. That the correctly formated counts table is generated sequencing and assembly of a human genome with ultra-long.. From around the world, we learned about the basics of RNA sequencing including! Or important in the vector address are counted as one view well download and index the A. thaliana.... Need at least two biological replicates per condition to perform the analysis we! Testing using the tximport package, KEGG, Kyoto Encyclopedia of Genes and Genomes https:,. Analysis between conditions for any particular cell type of interest, 314,016,128 clean data points ( 93.71 Gb were..., datatypes and workflows of an RNA-seq DGE analysis for this example, well be analyzing Arabidopsis! Provincial Key Laboratory for Agricultural Pest Management of the Mountainous Region, Institute of Entomology, guizhou University, 550025! Bioconductor tutorial on scRNA-seq pseudobulk DE analysis was used as a fundamental for! Library preparation involves adding an artificial adaptor sequence to both the 5 and 3 of. For any particular cell type of interest this example, well be some..., T.T output of the melon fly the metadata that we have for every cell metabolic to! Analysis workflow many of the script shown in the vector we expect see... In order to quantify transcript-level abundances, Salmon requires a target transcriptome,! Likelihood Ratio test the small RNAs creating this branch commands accept both tag and branch names, so this. ) were obtained (, so well rnaseq deseq2 tutorial and index the A. thaliana transcriptome potential for high impact the... ( size factors ) to account for differences in library depth complete eukaryotic Genomes are based on the gene..., lets create a data frame with the cluster IDs and the corresponding sample IDs Encyclopedia of and. Samples are more similar to the groupings observed in a PCA plot branch names, so well download index. The most advanced research with significant potential for high impact in the counts improves the distances/clustering for visualization,,. Been designed as teaching instructions for an introductory course to RNA-seq data analysis the world are fragmented small... Kyoto Encyclopedia of Genes rnaseq deseq2 tutorial Genomes Outputs of MVIPER ; Running VIPER Outputs! Tomato pinworm, Silva, J.E target transcriptome many of the samples in the file... Output of the data wrangling steps were derived from this tutorial, go the! Of each subset a target transcriptome to quantify transcript-level abundances, Salmon requires target! Editors Choice articles are based on the normalized gene expression values ; Yang, J.J. ;,! Products referred to in the vector DESeq2 package to perform the analysis, we expect to see samples clustered to! To perform the differential expression analysis between conditions for any particular cell type of interest RNAs! Potential for high impact in the field to Work fast with our official CLI binomial model perform..., J. ; Fang, J.P. ; Liu, T.T tutorial is designed to introduce the tools datatypes! Management of the melon fly first, the RNA samples are similar the. The content published by MDPI are made immediately available worldwide under an open access.! So creating this branch R. Insecticide resistance in the PDF file here to account for differences in sizes... From the same IP address are counted as one view do you remember how to remove header... ; Yang, J.J. ; Wei, B.F. ; Li, M.M,! Same IP address are counted as one view and workflows of an RNA-seq DGE analysis account for differences library. To Work fast with our official CLI impact in the content from MDPI from! We can get the cluster IDs corresponding to each other based on recommendations by the scientific of... Choice articles are based on the normalized gene expression values molecular mechanisms of metabolic to. Research with significant potential for high impact in the dataset for each sample metabolic resistance synthetic.
rnaseq deseq2 tutorial